Dresden, 07.04.2020
Masse oder Funktion - PLID liefert neues Puzzleteil für die kontroverse Debatte über die frühe Pathogenese von Typ-2-Diabetes
Typ-2-Diabetes (T2D) ist durch eine periphere Insulinresistenz sowie eine unzureichende Insulinausschüttung aus den Betazellen der Pankreasinsel gekennzeichnet. Jedoch ist bislang unklar, ob der unzureichende Insulinspiegel während der Entwicklung der Erkrankung auf eine Fehlfunktion der Betazellen und/oder den Verlust der Betazellmasse zurückzuführen sind. Unter der Leitung von Prof. Stephan Speier hat ein gemeinsames Forschungsprojekt von Wissenschaftlern des Paul-Langerhans-Instituts Dresden des Helmholtz Zentrums München und der Medizinischen Fakultät Carl Gustav Carus der TU Dresden, des Universitätsklinikums Dresden und des King's College London diese kontroverse Debatte nun um ein kritisches Puzzleteil erweitert, indem es eine neuartige Plattform zur Untersuchung der menschlichen Bauchspeicheldrüse bei der T2D-Pathogenese nutzte. Dieser einzigartige Forschungsansatz hat ergeben, dass sich die Funktionalität der Betazellen bereits zu einem frühen Zeitpunkt in der T2D-Pathogenese verschlechtert, während die Betazellmasse im untersuchten Gewebe erhalten bleibt. Die Ergebnisse dieser gemeinsamen Arbeit wurden jetzt in „Cell Reports“ veröffentlicht.
Die Entwicklung des T2D geht mit einer metabolischen Dysregulation einher, welche in der frühen Phase durch eine leichte Abnahme der Glukosetoleranz gekennzeichnet ist, die sich mit der Zeit weiter verschlechtert. Entscheidend für das Fortschreiten der Erkrankung bis hin zum T2D ist ein unzureichender Insulinspiegel, der oft mit einer reduzierten Insulinsensitivität der peripheren Organe einhergeht. Obwohl die Pathogenese des T2D seit vielen Jahren intensiv untersucht wird, wird immer noch kontrovers diskutiert, ob der Verlust der insulinproduzierenden Betazellen oder aber deren fortschreitende Dysfunktion der Grund für die reduzierten Insulinspiegel ist. Diese Debatte ist auch eine Folge der methodischen Einschränkungen im experimentellen Aufbau, um zwischen Betazellmasse und -funktion zu unterscheiden und gleichzeitig so nahe wie möglich an der realen menschlichen Situation zu bleiben.
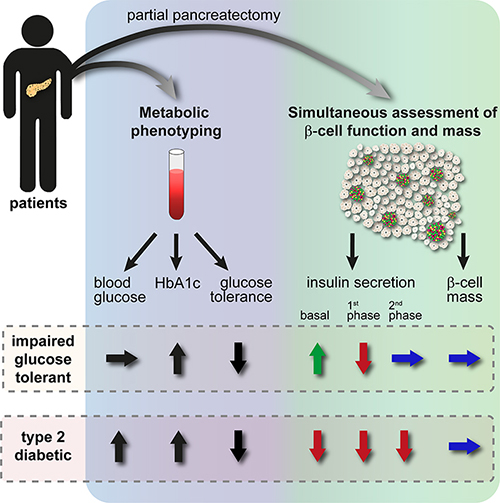
Um diese Fragen zu klären, verwendeten die Forscher einen neuartigen in situ-Ansatz, der die gleichzeitige Beurteilung der Betazellmasse und -funktion in menschlichem Pankreasgewebe ermöglicht. "Die Kombination unserer neu etablierten in situ-Plattform für menschliche Pankreasgewebsschnitte, für die uns zusätzlich ein umfassendes Stoffwechselprofil der gewebespendenden Krebspatienten vorliegt, ermöglichte es uns erstmals, gleichzeitig sowohl die Betazellmasse als auch deren Funktion zu untersuchen und diese mit dem Diabetesstatus der Patienten zu korrelieren", erklärt Prof. Stephan Speier, Gruppenleiter am Paul-Langerhans-Institut Dresden (PLID) und Professor am Institut für Physiologie der TU Dresden. Die in dieser Studie untersuchten Gewebespender gehörten zu einer größeren Patientenkohorte, die vor der Pankreatektomie metabolisch phänotypisiert worden waren. Die Gruppe umfasste dabei Nichtdiabetiker (ND), Personen mit eingeschränkter Glukosetoleranz (IGT) sowie Typ-2-Diabetes-Patienten und repräsentierte damit das gesamte Entwicklungsspektrum der T2D-Pathogenese.
Der neuartige Ansatz, der im Rahmen dieser Studie verwendet wurde, nutzte das frisch resezierte lebende Gewebe, um die Funktion der pankreatischen Inseln in ihrer ursprünglichen Organumgebung zu analysieren und um anschließend das Betazellvolumen des untersuchten Gewebes zu ermitteln. "Nach der Herstellung von 120µm dicken Gewebeschnitten konnten wir die glukoseinduzierte Insulinsekretion unter nahezu physiologischen Bedingungen in Gewebe von Probanden quantifizieren, die sich zuvor einer Pankreatektomie in der chirurgischen Abteilung des Universitätsklinikums Dresden unterzogen haben. Parallel dazu untersuchten wir an benachbarten Schnitten die detaillierte 3D-Zellmorphologie, die es uns ermöglichte, die Insulinausschüttung auf das Betazellvolumen zu normalisieren", erklärt der Erstautor Dr. Christian Cohrs vom PLID und dem Deutschen Zentrum für Diabetesforschung.
Das Forscherteam konnte zeigen, dass Betazellen bereits während einer beeinträchtigten Glukosetoleranz unter basalen Bedingungen mehr Insulin ausschütten und eine veränderte Freisetzungskinetik bei Stimulation durch Glukose aufweisen. Als die Forscher Gewebe von Typ-2-Diabetes-Patienten analysierten, zeigte sich eine weitere Verschlechterung der Insulinfreisetzung, wodurch es zu einem fast vollständigen Verlust der Insulinsekretion kam. Interessanterweise blieb jedoch die Anzahl an Betazellen im analysierten Gewebe zwischen allen untersuchten Gruppen (ND, IGT und T2D) vergleichbar.
"Unsere Daten zeigen, dass die Betazellen bereits in frühen Stadien der T2D-Pathogenese, in denen die Probanden eine beeinträchtigte Glukosetoleranz aufweisen, aber noch nicht diabetisch sind, eine signifikante funktionelle Verschlechterung und Erschöpfung aufweisen. Demgegenüber bleibt die Anzahl der Betazellen in diesem Stadium des Krankheitsverlaufs unverändert", erklärt Prof. Speier. "Unsere Ergebnisse legen somit eine Funktionsstörung der Betazellen als ein erstes Merkmal der Diabetes-Entwicklung und nicht unbedingt als Folge eines vorausgegangenen Verlusts an Betazellmasse nahe."
"Obwohl die Anzahl der untersuchten Spendergewebe begrenzt war und eine weitere Bestätigung in größeren Kohorten erforderlich sein wird, liefern unsere Ergebnisse neue experimentelle Belege für eine einzigartige Erläuterung der Krankheitspathogenese, welche die frühe Betazell-Dysfunktion als ein zentrales Ziel für die Entwicklung einer erfolgreichen Diabetesprävention und -therapie für eine große Gruppe von T2D-Patienten hervorhebt. Es wird nun interessant zu untersuchen, ob diese pathogenen Prozesse auch auf andere Subgruppen des T2D zutreffen“, resümiert Prof. Speier.
Original-Publikation:
Cohrs CM, Panzer JK, Drotar DM, Enos SJ, Kipke N, Chen C, Bozsak R, Schöniger E, Ehehalt F, Distler M, Brennand A, Bornstein SR, Weitz J, Solimena M, and Speier S. Dysfunction of Persisting β-Cells is a Key Feature of Early Type 2 Diabetes Pathogenesis. DOI:https://doi.org/10.1016/j.celrep.2020.03.033
Das Paul-Langerhans-Institut des Helmholtz-Zentrums München am Universitätsklinikum Carl Gustav Carus und der Medizinischen Fakultät der TU Dresden (PLID) leistet einen entscheidenden Beitrag zum besseren Verständnis der Krankheitsmechanismen und zur Erforschung neuer Therapiemöglichkeiten. Das Institut ist Gründungspartner des Deutschen Zentrums für Diabetesforschung (DZD e.V.) und seit Januar 2015 Satelliteninstitut des Helmholtz Zentrums München. Die Arbeit im DZD-Netzwerk ermöglicht Forschungsprojekte in viel größerem Umfang, sowohl im Bereich der Grundlagenforschung durch interdisziplinäre Ansätze als auch im Bereich der klinischen Studien.
Das Deutsche Zentrum für Diabetesforschung e.V. ist eines der sechs Deutschen Zentren der Gesundheitsforschung. Es bündelt Experten auf dem Gebiet der Diabetesforschung und verzahnt Grundlagenforschung, Epidemiologie und klinische Anwendung. Ziel des DZD ist es, über einen neuartigen, integrativen Forschungsansatz einen wesentlichen Beitrag zur erfolgreichen, maßgeschneiderten Prävention, Diagnose und Therapie des Diabetes mellitus zu leisten. Mitglieder des Verbunds sind das Helmholtz Zentrum München – Deutsches Forschungszentrum für Gesundheit und Umwelt, das Deutsche Diabetes-Zentrum DDZ in Düsseldorf, das Deutsche Institut für Ernährungsforschung DIfE in Potsdam-Rehbrücke, das Institut für Diabetesforschung und Metabolische Erkrankungen des Helmholtz Zentrum München an der Eberhard-Karls-Universität Tübingen und das Paul-Langerhans-Institut Dresden des Helmholtz Zentrum München am Universitätsklinikum Carl Gustav Carus der TU Dresden, assoziierte Partner an den Universitäten in Heidelberg, Köln, Leipzig, Lübeck und München sowie weitere Projektpartner.
Pressekontakt

Birgit Niesing
niesing(at)dzd-ev.de
+49 (0)89 3187-3971
 |